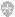偏头痛和脑卒中的互相关联
2014-09-21 10:59:23 来源: 丁香园 作者: 评论:0 点击:
丁香园 作者: 评论:0 点击:
近期 Headache 杂志发表综述,阐明了偏头痛与脑卒中之间的关系。偏头痛可增加卒中的风险;血管病变、卵圆孔未闭、肺动静脉畸形、动脉壁的损伤(如急性动脉夹层)可出现偏头痛先兆发作或卒中;两者之间的机制或与大脑超兴奋以及播散性去极化的易感性增加有关。
偏头痛为慢性间歇性头痛,发作持续时间为 4~72 小时,重度的单侧头痛常表现为搏动样,伴恶心、畏光和 / 或畏声。1/3 的偏头痛患者存在先兆神经系统症状,先兆期约为头痛发作前 1 小时。偏头痛的发病率在女性中为 43%,男性为 18%;但在青年期之前男女间发病率相似,均为 4%。偏头痛为导致残疾排名前 5 位的疾病,根据 2010 年全球疾病负担研究,可引起神经残疾全球负担的 50%。
根据世界卫生组织的统计,脑卒中是全球排名第三的致死性疾病(在全部死亡事件中占 9%),脑卒中可引起长期残疾,占健康支出费用的 2%~4%。在发展中国家脑卒中的死亡率为 50~100/10 万人 / 年,每年约有 80 万脑卒中患者,其中 60 万为新发患者。虽然脑卒中男性发病率高于女性;但在低于 35 岁和高于 75 岁的人群中,女性发病率高于男性。
脑卒中的风险因子分为可改变性风险(如高血压、糖尿病和吸烟)或固定性风险(如男性、高龄)。然而,与缺血性心脏病不同,风险因子仅能解释 60% 的卒中发病,而前者超过 90% 的发病可被风险因子所解释。因此,需要进一步确定和改变脑卒中新的风险因子(如偏头痛)。
一、偏头痛和脑卒中的临床联系
流 行病学研究表明,偏头痛(尤其是先兆型)和脑卒中(缺血性和出血性)之间确有联系。临床和人群研究显示,偏头痛可能是令缺血性脑卒中风险加倍的独立因素, 在年轻健康的先兆型偏头痛患者中尤其如此;女性偏头痛缓则的脑卒中风险显著增加,而口服避孕药的偏头痛女性脑卒中风险增加至 9 倍。偏头痛患者增加的卒中风险,与偏头痛和血管疾病的遗传因素(如亚甲基四氢叶酸还原酶或血管紧张素转化酶基因的多态性)有关。
而神经影像学研究表明偏头痛与缺血性脑卒中之间存在联系,偏头痛患者白质异常或梗死样病灶的发生率较高(总体比值比 1.68);有趣的是,在一些研究中,白质高密度的程度与偏头痛发作的频率正相关,提示着两者之间的剂量 - 反应关系。
80% 的卒中为缺血性,而 20% 为出血所致;最近一项纳入 1600 名出血性脑卒中患者的 meta 分析显示,先兆型偏头痛亦可增加出血性脑卒中(蛛网膜下出血和颅内出血)的风险。
二、脑的超兴奋:偏头痛和脑卒中的共同联系
偏头痛和脑卒中的因果关系尚未完全确定。实验研究提示,偏头痛可能诱导播散性去极化(spreading depolarization,SD),令脑部易受缺血性脑卒中的损害。
SD 可能为先兆型偏头痛和脑卒中的共同机制,因 Na+/K+-ATP 酶活性受损,阳离子外流减少、内流增加或血管病变相关的缺血性损伤,引起脑部超兴奋所致。在啮齿类动物中,脑部小动脉的微血栓梗塞可诱发 SD;在临床上,女性先兆偏头痛患者的外周循环中可检测到内皮微粒。SD 的检测因其较好的阳性阴性预测值,可作为临床前模型用于筛查偏头痛药物。
SD 可被多种因素诱发(图 1),常见的为 KCL、电或机械刺激。最近,Baca 等人的研究显示,SD 还可被非侵入性光刺激和星形胶质细胞的选择性激活所诱导。SD 时,离子梯度被一过性破坏,且神经元膜阻抗并联分流、电活性丧失、神经元肿胀伴随棘突变形。
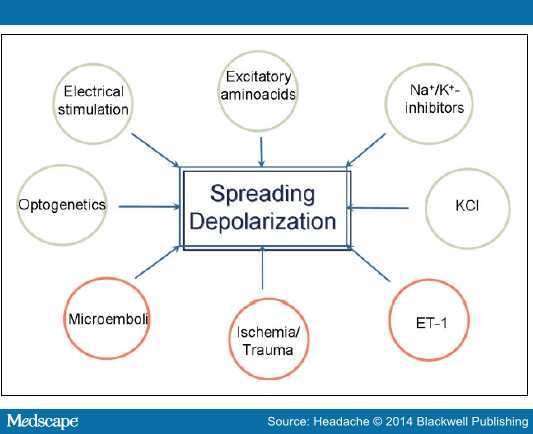
图 1. SD 的触发包括兴奋性(灰色圆圈)和血管性因素(红色圆圈)。细胞外 K+ 浓度升高是重要事件;小鼠实验表明脑动脉的微栓子可引起 SD,提示 SD 可能为偏头痛与脑卒中的共同联系。
在短暂充血 2 分钟后,SD 可减少脑部血流长达 2 小时;充血可为神经元提供养分,帮助神经元从 SD 诱发的离子和水的失衡中恢复。随后,在长时间的缺血状态下,血管反应和功能性激活均下降。这些由 SD 所引起的不同血管运动的程度和时程,依赖于多种因素。
与脑卒中的病理类似,通过星形胶质细胞钙离子水平、细胞外 K+ 浓度、大电导 Ca2+ 激活的 K+ 通道(BK 通道)活性的改变,血管运动反应逐渐变为重度的血管收缩。
在蛛网膜出血的动物脑片模型中,当围绕脑血管的星形胶质细胞足突内钙离子浓度超过 500 nmol/L 时,通过 BK 通道介导的 K+ 外流,可引起平滑肌去极化和血管收缩。相反,星形胶质细胞足突钙离子浓度中度增高即可触发扩血管物质的释放。BK 通道活性的增加,刺激了小动脉内向整流钾通道(Ki)的活性,令脑实质内动脉的平滑肌超极化,进一步促进了血管扩张。
动物和人类实验均低估了偏头痛和脑卒中中 SD 的重要性(图 2)。

图 2. 大脑超兴奋或为偏头痛和脑卒中的共同联系,神经血管单元的动态相互作用是偏头痛和脑卒中重要的病理生理学改变。近期研究显示,反映大脑超兴奋的 SD,不仅参与偏头痛的发病机制,亦可增加偏头痛患者的卒中风险。
1. 偏头痛的播散性去极化
在 Leao 首次描述 SD 的 3 年前,Lashley 推测其偏头痛的视觉前兆为一种沿着视皮质传播的电生理现象,速度为 3 mm/ 分钟。1958 年,Milner 认为 Lashley 根据视网膜皮层投射所推测的视觉前兆就是 SD。2001 年,Hadjikhani 的研究证实 SD 在偏头痛前兆中扮演了重要的角色。
偏头痛患者视觉先兆时,高场强功能性 MRI 显示,血氧水平依赖的信号初始稍有增强,之后持续下降,反映出神经元活动的先增加后抑制;该信号的改变始于枕极,传播的速率与 SD 相似;提示 SD 和偏头痛受相同的因素调节。
男性性激素以及偏头痛的预防药物可抑制 SD 和偏头痛,而女性性激素可增加 SD 的易感性。偏头痛和 SD 也有共同的触发因素,如脑部小动脉的微栓子栓塞可引起 SD、偏头痛甚至卒中。
SD 也参与头痛。Moskowitz 和 Kraig 的研究显示,SD 可引起三叉神经脊束核尾侧亚核神经元活性标记物 cFos 的上调,而 cFos 参与着头痛的伤害感受器通路。近期的研究表明,在 30 分钟的潜伏期后,SD 可激活脑膜的伤害感受器和三叉神经脊束核尾侧亚核内的三叉神经血管纤维,这与临床上头痛的先兆十分相似。
2. 脑卒中的播散性去极化
SD 亦可见于卒中的缺血半暗带中。在脑缺血起始时,Na+/K+-ATP 酶逐渐失活可引起细胞外 K+ 水平缓慢上升,直至达到 SD 的诱发阈值。SD 通过刺激 O2 和葡萄糖的消耗,加剧了缺血半暗带内的代谢失衡;通过血管的收缩作用恶化了脑组织内的血液灌注。
缺血诱导了脑组织自体平衡的改变,如 NO 下降,K+ 浓度倒置;因此在 SD 诱导电化学功能障碍后,缺血促进了神经元的损伤。组织越缺血,则血管收缩反应越严重,神经 - 血管之间的偶联越弱。
在实验环境下,高血氧或高血糖均不能恢复血管反应,提示大脑灌注压可能较组织的能量状态更为重要。当神经元从 SD 后存活后,不能复极化的神经元可发生死亡。在大鼠中,重度缺血可导致皮质神经元和星形胶质细胞广泛坏死,而人工诱导的 SD 可逐渐加重坏死,提示神经元去极化可引起细胞死亡的级联反应,就算从离子失衡中恢复也不代表神经元具有活性。
在硬脑膜下放置电极的电生理研究肯定了 SD 在脑损伤或卒中患者中反复发生,但缺血诱导的 SD 在健康组织中则难以与正常 SD 相区分。缺血性 SD 与脑卒中患者的神经功能恶化有关,可加速代谢失衡,引起非缺血性组织的继发性损伤。
3. 播散性去极化为偏头痛和脑卒中的共同联系
动物实验和人群研究提示,对 SD 易感性增加可令偏头痛患者易受缺血和卒中风险的损害。有偏头痛史的出血性卒中患者,对缺血性神经损伤和死亡的风险增高。表达人神经性(家族性偏瘫型偏头痛)或血管性(伴有皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病,CADASIL))偏头痛基因突变的小鼠,存在高频率的 SD、动脉阻塞、卒中结局更差。
家族性偏瘫型偏头痛 1 型(FHM1)由 Cav2.1 通道所致,该通道可将去极化激活的突触前 Ca2+ 内流放大,通过降低 Cav2.1 开发电压和延迟通道的失活增加谷氨酸的释放。FHM1 转基因小鼠对 SD 的反应与临床上患者的先兆症状类似;对 SD 的易感性增加。大脑中动脉阻塞后,FHM1 小鼠的缺氧性去极化出现的更早,表现出连续性缺血诱导的 SD。
增加的突触前 Ca2+ 内流和兴奋性神经递质谷氨酸的连续释放或可解释 FHM1 小鼠快速出现且数量增多的 SD;而 FHM1 小鼠低灌注区域的扩大与 SD 的频率相关;每次 SD 出现时,缺血半暗带向梗死区融合,梗死面积不断增加,结果在弥散加权 MRI 上可见超急性梗死的加速进展。。
FHM1 小鼠梗死区边缘的皮质血流较高,提示组织的生存阈值增高(图 3),梗死区脑组织血流减少至缺血前基线指的 45%;而野生型小鼠梗死组织血流减少至 34%。
血管性 Notch3 R90C 基因变异(可引起人偏头痛相关综合征 CADASIL)的小鼠,可出现相似的、重度卒中的表现。与 FHM1 小鼠相似, TgR90C 小鼠对 SD 的易感性较高,缺氧性去极化可早期出现,缺血性去极化的频率增加,梗死面积增大,神经系统结局恶化;这些结果提示在疑似偏头痛的脑内,脑实质的病变机制(如组织去极化)对卒中风险的增加起着重要的作用。
图 3. FHM1 基因变异小鼠脑组织生存阈值的增加。在梗死 48 小时候,采用激光散斑对比成像反映脑血流(CBF)的变化。FHM1 小鼠平均组织生存阈值高于野生型小鼠(P = .048),提示前者更易受缺血的损害,需要更多血流,组织才能存活。*P <0 .05 。
全基因组关联研究显示,常见类型的偏头痛均有脑兴奋性的增加。近期一项研究比较了脑卒中患者弥散加权成像 MRI,发现:与无偏头痛史脑卒中患者的相比,有偏头痛史的脑卒中患者更年轻,女性居多,梗死多由于心源性栓子。偏头痛与小的皮质梗死相关,提示在偏头痛的大脑中,针对小的缺血性事件,组织的生存阈值增加。
其它研究进一步证实了大脑超兴奋可增加脑缺血的易感性。譬如,癫痫患者发病年龄超过 60 岁也与脑卒中风险增高相关。在临床环境中证实动物实验,将有助于确定偏头痛为快速梗死进展的风险因素,而缺血半暗带的加速丧失可意味着偏头痛患者急性卒中干预的治疗窗更为短暂。
大脑超兴奋可解释目前文献中关于偏头痛与卒中关系的三种形式。
(1) 偏头痛与脑卒中之间存在共同的病因。系统性分析提示,卵圆孔未闭与偏头痛之间存在相关(比值比 2.5,95% 可信区间 2.0~3.1),且与缺血性卒中之间亦有关联(比值比 2.9,95% 可信区间 2.1~4.0)。微栓子通过心脏的右向左分流进入脑部血液循环,引起小动脉梗塞(反常栓塞),导致脑缺血和诱发 SD/ 偏头痛,倘若重度缺血还可发生卒中。
有国际间研究(非盲非随机)显示,隐源性缺血性卒中和难治性偏头痛可从经皮封堵卵圆孔未闭的治疗种获益,但尚无随机对照研究证实其中的关系。
(2) 原发性脑缺血性事件,如动脉夹层,可加速或诱发偏头痛发作,偏头痛与颈动脉夹层之间的机制或与临床上颈动脉夹层患者循环中“沉默的”微栓子有关,而后者可 引起缺血和诱发 SD/ 偏头痛发作。据报道,自发性颈动脉夹层的卒中患者,偏头痛发生率增高;而 TGFβR2 基因突变引起的家族性主动脉夹层的 14 名患者中, 其中 10 名患有偏头痛。
但上述多数研究存在回顾性设计、样本量较小等缺点,最近一项病例对照研究,通过分析多中心的“颈动脉夹层与缺血性脑卒中”的研究数据,肯定了偏头痛更常见于颈动脉夹层的患者。
(3) 偏头痛作为缺血性卒中的直接诱因时,称为“偏头痛性脑梗死”,是罕见的卒中亚型,在年轻患者中占全部缺血性梗死的 20%,女性发病率为其它卒中的 2~3 倍,主要累及后循环;其症状持续超过 60 分钟,神经影像上可见相关脑区的缺血性梗死;临床症状与其它卒中类型相异,发病前伴有典型的先兆。动物实验提示,转基因的偏头痛小鼠 SD 诱导的血流减少更为严重;且枕叶是最易发生 SD 的皮质区域。
三、临床意义
本综述假设,偏头痛相关的大脑超兴奋,可增加对脑缺血的易感性。倘若该假设被临床试验证实,则应治疗 / 预防偏头痛患者(尤其是高卒中风险的患者)的大脑超兴奋。
很多药物可影响 SD 的发生,譬如一些麻醉和镇静药,可影响神经元兴奋依赖性的离子稳态,对 SD 产生急性抑制。近期一项国际间多中心的回顾性分析显示,抗兴奋性 N- 甲基 -D- 天冬氨酸(NMDA)受体拮抗剂氯胺酮可抑制创伤性脑损伤、蛛网膜下腔出血或卒中患者的 SD 及簇状 SD(≥3 SDs/3 小时)。
类似的, NMDA 拮抗剂 MK-801 也可抑制缺血诱导的 SD,对 FHM1 基因突变小鼠的卒中有保护作用;与野生型小鼠相比,采用 MK-801 预处理的 FHM1 小鼠,卒中结局减轻。
虽然在动物实验中抑制 NMDAR 相关通路可保护卒中,但数个大规模的临床试验都未能在患者
上一篇:临床综述:老年人持续性疼痛管理
下一篇:NEJM 指南:老年抑郁




")

临床要点")


频道总排行
医学推广

频道本月排行
热门购物
评论排行
- 2011年临床执业医师考试实践技能真...(13)
- 腋臭手术视频(12)
- 2008年考研英语真题及参考答案(5)
- 节食挑食最伤女人的免疫系统(5)
- 核辐射的定义和单位(5)
- CKD患者Tm与IMT相关(5)
- 齐鲁医院普外科开展“喉返神经监护...(5)
- windows7激活工具WIN7 Activation v1.7(5)
- 正常微循环(5)
- 美大学性教育课来真的 男女上阵亲...(4)